


Necroptosis
Classically, necrosis and apoptosis have been described as separated and opposite entities by the fact that the hallmark of necrosis is the generation of inflammation caused by the uncontrolled release of the cellular debris in the micro-environment, while apoptosis, as well as the other forms of Programmed Cell Death, avoid inflammation by means of integrated biochemical steps. However, recently, in-depth investigations showed that in certain settings also necrotic death can occur through pre-definite molecular pathways, while certain key steps of Programmed Cell Death can result in a necrosis-like death. So, new terms have been coined: necroptosis or programmed necrosis. These terms describe cases whereby necrosis is not accidental, but rather regulated and programmed, being therefore distinct from both apoptosis and accidental necrosis. The first evidence raised from the observation that Tumor Necrosis Factor-α (TNF-α) is able to induce both apoptosis and necrosis, especially in the presence of caspase-inhibitors. TNF-α binds specific receptors on the cell surface that transduce death signals via the extrinsic pathway of apoptosis, in other words by activating caspase 8. However, when caspase 8 is blocked by synthetic or viral inhibitors, or by drastic depletion of energy (ATP depletion), there is a shift from apoptosis toward necrosis, mediated by the RIP kinases, RIPK1 and RIPK3. Once activated, the RIPK1-RIPK3 complex stimulates BAK and BAX to generate massive Mitochondrial Outer Membrane Permeabilization (MOMP), with subsequent overwhelming production of Reactive Oxygen Species, responsible for diffuse oxidation of cellular component and eventually of lytic damages. Release of cellular contents and of Damage-Associated Molecular Patterns (DAMPs) occurs, which triggers activation of the immune system and initiates inflammation. Thus, these events are not passive, like in the traditional necrosis, but rather induced by an intracellular, dedicated signaling system, represented by the RIP kinases, RIPK1 and RIPK3.
Necroptosis has been described in the context of severe ischemia-reperfusion conditions, thereby participating in the pathogenesis of important diseases. In fact, myocardial, neuronal, and kidney necroptotic damage, strictly depending on RIPK1/RIPK3 activity, occurs when intense oxygen/nutrients deprivation takes place, e.g., in the territories directly perfused by an artery that suddenly plugs.
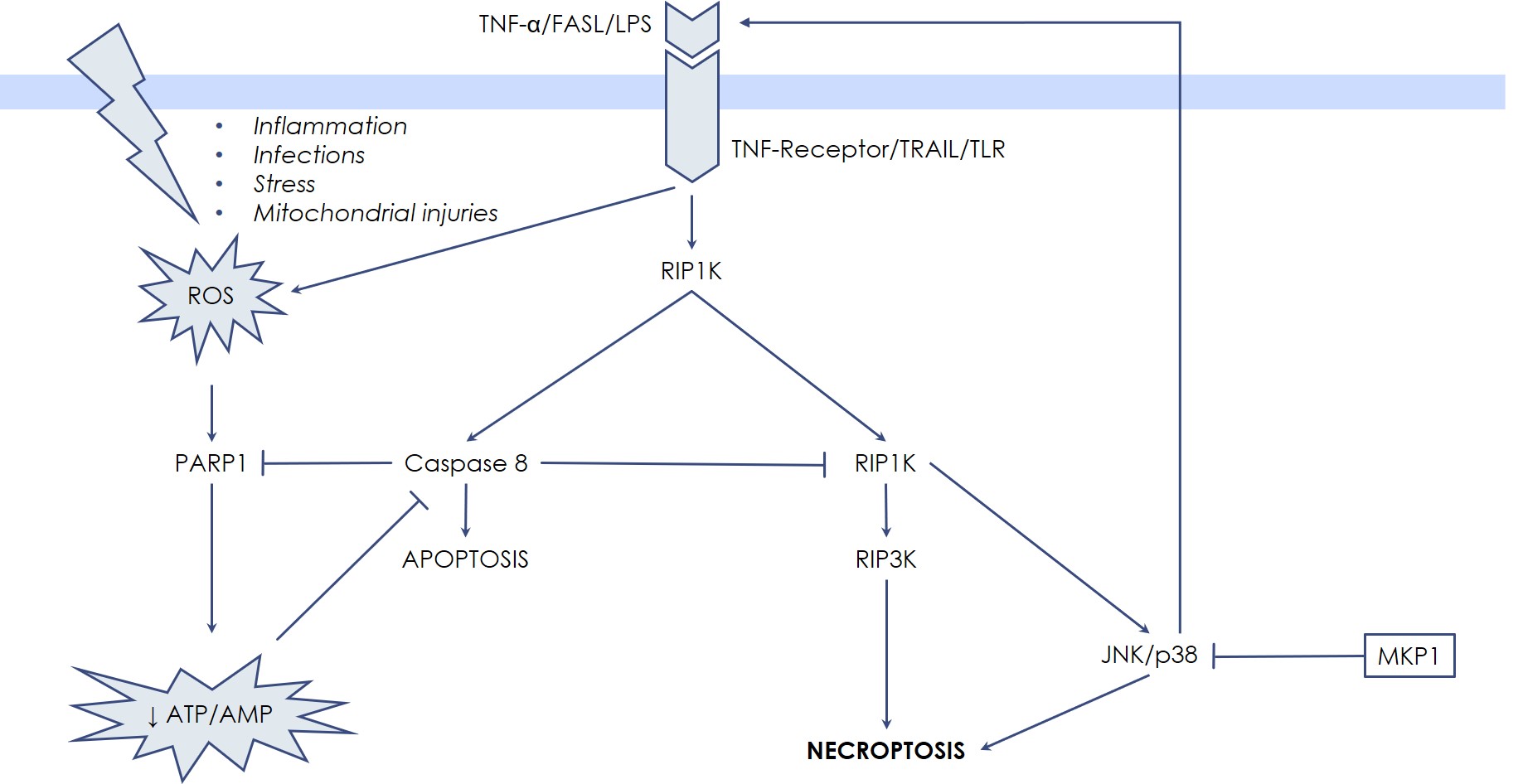
In this case the unexpected nutrients deprivation (ATP depletion), together with ROS elevation, drives the necrotic process through the direct stabilization of RIPK1-RIPK3 complex, which has JNK and p38 MAPK as downstream targets. For these reasons, it is possible to interfere with the necrotic death process, through modulation of JNK and p38 MAPK activity by Udonitrectag, that primarily acts via fast induction of MKP1 expression. Now substantial experimental work demonstrates the involvement JNK and p38 MAPK in the necrotic process. For example, Rotenone (a poison used to reproduce the pathogenesis of some neurodegenerative diseases) induces necroptotic death via JNK and p38 MZAPK activity in retinal ganglion cells; similarly, microglia (resident immune cells in the central nervous system) undergoes necroptotic death mediated by JNK when caspase 8 is blocked.